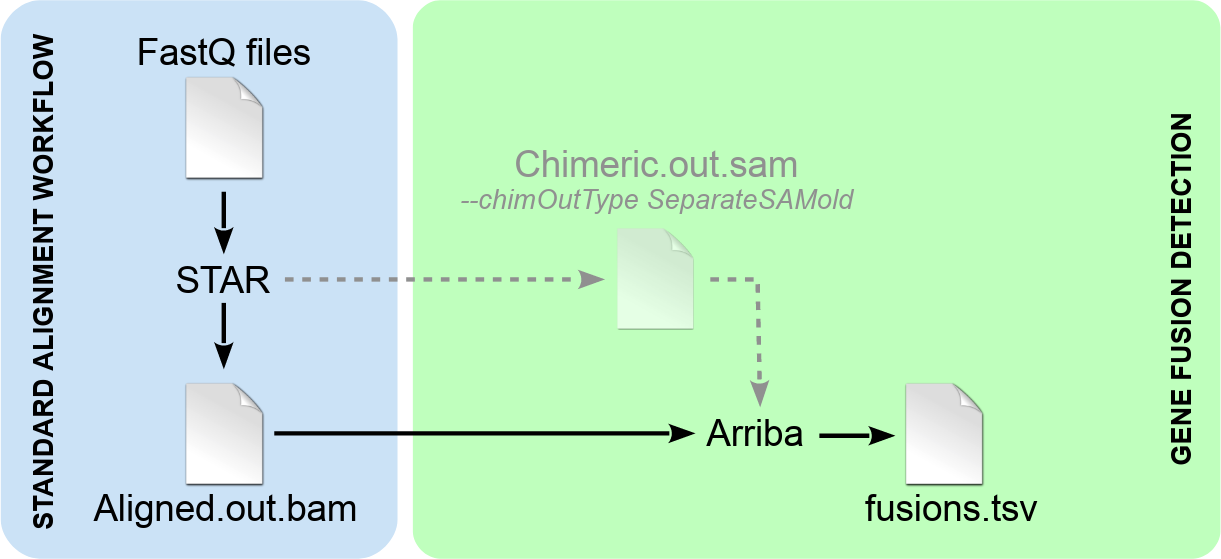
Fusion detection with Arriba is based on the STAR aligner. It is an extension of the regular alignment workflow, which can be incorporated with few modifications. The addition of Arriba does not affect the normal alignments. The workflow yields fusion predictions as well as normal alignments that can be used for other downstream analyses such as expression quantification or variant calling. Like so, fusion detection incurs negligible computational overhead, since it adds only a few minutes of runtime to the regular alignment workflow.
Implementing Arriba is done in two steps:
-
Detection of chimeric reads must be enabled in STAR by specifying the parameter
--chimSegmentMin. In addition, the parameter--chimOutType WithinBAMmust be specified to cause STAR to report chimeric reads as supplementary alignments in the main output fileAligned.out.sam. Old versions of STAR (or when STAR is run with--chimOutType SeparateSAMold) wrote supplementary alignments to a separate file namedChimeric.out.sam. Arriba is compatible with this mode of use (see parameter-c), but it is deprecated, because STAR might not support it anymore in the future. -
Arriba extracts the supplementary alignments from the given input file(s). The supplementary alignments represent evidence about translocations, inversions, duplications, and deletions larger than the usual intron size (as defined by the parameter
--alignIntronMax). In order to find fusions arising from deletions smaller than the maximum intron size, Arriba also extracts alignments which cross the boundaries of annotated genes. Once all alignments have been extracted, it applies a set of filters to remove artifacts and transcripts observed in healthy tissue. The final output is a list of fusion predictions which pass all of Arriba's filters.
Demo script
Arriba comes with a script run_arriba.sh that illustrates the usage of all components of the workflow and how they are meant to interact with each other. The script is deliberately kept simple and lacks error checking, input validation, adapter/quality trimming etc. The script is meant only as a guide which demonstrates how to integrate Arriba into your own STAR-based workflow. The following paragraphs explain the steps of the demo script:
In order for STAR to search for chimeric alignments, the parameter --chimSegmentMin must be specified and the parameter --chimOutType must be set to WithinBAM or SeparateSAMold. In addition, the following parameters related to chimeric alignment are recommended for improved sensitivity (requires STAR version 2.7.6a or higher):
--outFilterMultimapNmax 50 \
--peOverlapNbasesMin 10 \
--alignSplicedMateMapLminOverLmate 0.5 \
--alignSJstitchMismatchNmax 5 -1 5 5 \
--chimSegmentMin 10 \
--chimOutType WithinBAM HardClip \
--chimJunctionOverhangMin 10 \
--chimScoreDropMax 30 \
--chimScoreJunctionNonGTAG 0 \
--chimScoreSeparation 1 \
--chimSegmentReadGapMax 3 \
--chimMultimapNmax 50
Arriba does not care if the BAM files are sorted/indexed or not. For maximum speed, STAR's output should be piped directly to Arriba, such that chimeric reads are extracted while STAR is still running. STAR can be instructed to write alignments to STDOUT using the parameters --outStd BAM_Unsorted and --outSAMtype BAM Unsorted. Furthermore, it is recommended to pipe the alignments in uncompressed format to Arriba (--outBAMcompression 0), because BAM compression is not parallelized well in STAR and becomes a bottleneck when using more than six to eight threads.
A complete call of STAR in conjunction with Arriba may look like this:
STAR \
--runThreadN 8 \
--genomeDir /path/to/STAR_index --genomeLoad NoSharedMemory \
--readFilesIn read1.fastq.gz read2.fastq.gz --readFilesCommand zcat \
--outStd BAM_Unsorted --outSAMtype BAM Unsorted --outSAMunmapped Within --outBAMcompression 0 \
--outFilterMultimapNmax 50 --peOverlapNbasesMin 10 --alignSplicedMateMapLminOverLmate 0.5 --alignSJstitchMismatchNmax 5 -1 5 5 \
--chimSegmentMin 10 --chimOutType WithinBAM HardClip --chimJunctionOverhangMin 10 --chimScoreDropMax 30 \
--chimScoreJunctionNonGTAG 0 --chimScoreSeparation 1 --chimSegmentReadGapMax 3 --chimMultimapNmax 50 |
arriba \
-x /dev/stdin \
-o fusions.tsv -O fusions.discarded.tsv \
-a /path/to/assembly.fa -g /path/to/annotation.gtf \
-b /path/to/blacklist.tsv.gz -k /path/to/known_fusions.tsv.gz -t /path/to/known_fusions.tsv.gz -p /path/to/protein_domains.gff3
Note: In this example, the same file is passed to the parameters -k and -t, because it is used for two purposes: applying sensitive filtering parameters to known fusions (-k) and tagging known fusions in the tags column (-t). However, it is possible to use different files for these two parameters if a user wants to separate the two tasks.
For a comprehensive list of the parameters, please refer to section Command-line options.